Inflammation, cancer, and NF-κB
Cancer isn't a single disease, and there are many possible things that can "cause" even a single type of cancer. However, a lot of things basically work in the same general way to promote cancer. One way that's been long suspected though poorly understood is tangled up in the process of inflammation.
At the very center of this story is a transcription factor called NF-κB. It's perhaps best known for its role in inflammation. But we discussed it in connection with cancer over two years ago, here. Since NF-κB is a transcription factor it affects the expression of many genes. Consequently, it's involved in many other biological phenomena, a few of which have been discussed here, here, and here.
Let's review some of the characteristics shared by most forms of cancer. One frequently cited summary, due to Douglas Hanahan and Robert Weinberg, names six "hallmarks of cancer" – self-sufficiency in growth signals, insensitivity to growth-inhibitory signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis. It turns out that inflammation and NF-κB can affect most of these hallmarks, but we'll eventually focus on the role NF-κB plays in inhibition of apoptosis (programmed cell death).
Before we get into details, let's go over the way that cancer typically develops. Each of the hallmarks represents a failure in one or more parts of the machinery of cells to properly regulate the cell's life cycle. The bad thing about cancer, of course, is that a certain group of cells – which might all be descendants of a single aberrant cell – begins to grow and proliferate in an unregulated way. Eventually large numbers of unregulated cells will cease to function as required in the organ where they reside – the brain, the liver, breast tissue, or whatever. Instead they will acquire the ability to migrate and take up residence in places where they continue to proliferate and then disrupt the proper function of whatever organ they wind up in. This is metastasis, and it is usually fatal to the animal in which it occurs.
The cell's machinery is ultimately controlled by the way specific genes of the cell's DNA are expressed to produce proteins. So the reason a cell initially becomes improperly regulated is usually the occurrence of fresh damage to the cell's DNA. It's true that problematic DNA mutations can be inherited from parents, such as the well-known breast cancer genes BRCA1 and BRCA2. Signifcantly, the normal function of both these genes, when not mutated, is to produce proteins whose job is to detect and/or repair DNA damage or to arrest cell proliferation if DNA damage is detected but can't be repaired. So mutated versions of these genes do not exactly "cause" cancer themselves, since some mutation to other genes that actively induce excessive cell proliferation is also required. The potential for cancer may simply remains latent for awhile in cells with mutated genes, regardless of whether the mutation was inherited or occurred much later in life.
But eventually uncorrected DNA damage will occur and affect genes that do induce excessive proliferation. Metastatic cancer eventually results, regardless of whether the first harmful mutations occur in genes that induce proliferation or genes that help regulate proliferation. Cells that contain such mutations divide more frequently and outcompete for resources cells that are functioning properly. So a Darwinian evolutionary scenario arises in which cells that are working "correctly" lose out to cells that become better "adapted" to the job of simply replicating themselves.
The point of this general discussion about cancer is to help clarify what it means to say that something "causes" cancer. In reality, there's never a single event that is the "cause". A number of things have to go wrong before some population of cells with similar defects is numerous enough to outcompete properly functioning cells and to go on to acquire further defects when the mechanisms that normally protect against damage themselves begin to fail.
There appear to be several ways in which NF-κB can contribute to the development of cancer, but one of the least surprising ways is the fact that NF-κB is able to inhibit apoptosis. This ability is not accidental. Most likely it is properly there to enable proliferation of immune system cells in the presence of an infection, as indicated by inflammatory signals.
To return to our main topic, inflammation is a thoroughly normal part of the immune system's operation. NF-κB plays an important part in the inflammatory process. But in simply doing their jobs, these things can, under the right conditions when other cellular mechanisms befome defective, also contribute to the development and progress of cancer.
Normally, the ability of NF-κB to inhibit apoptosis doesn't present a risk of cancer because NF-κB is regulated by other proteins, as we'll discuss later. However, if there are gene mutations that affect NF-κB regulatory proteins, the "safety catch" mechanism may be compromised, and apoptosis may be inhibited when it shouldn't be. Such a problem is more likely to occur when inflammation is present, since then the "safety catch" is already partly disabled.
Another mechanism that keeps NF-κB inhibition of apoptosis in check is the existence of pro-apoptotic proteins such as p53. (We've discussed p53 a number of times before, most recently here, where new research about the anti-cancer properties of p53 is described.) Such proteins are normally subject to regulation themselves, and they become available and active only when needed, such as when DNA damage is detected. However, when one of these proteins has itself been compromised by a gene mutation, inflammation and resultant NF-κB activity can inappropriately inhibit apoptosis, because pro-apoptotic factors are weakened or sidelined.
As it turns out, according to recent research, among the ways that p53 promotes apoptosis is by direct interference with NF-κB's ability to inhibit apoptosis. Further, certain mutations of the p53 gene can remove p53's pro-apoptotic ability, allowing inflammation and NF-κB to contribute to development of cancer. (See here. We'll discuss that in a separate article.)
The high-level view of all this is that cells exist continually in a state of balance between opposing possibilities. Pro-apoptotic and anti-apoptotic mechanisms are not only regulated independently, but they also keep each other in check. But when mutations in any number of possible genes upset the balance, otherwise normal and useful mechanisms can lead to cancer.
Now let's go a little deeper into the subject of various factors that actively contribute to cancer, starting with DNA damage – mutations. The class of things that cause mutations comprises factors such as carcinogenic chemicals, reactive oxygen species ("free radicals"), ultraviolet light, ionizing radiation (e. g. x-rays, radon gas), and some types of viruses. DNA is also at risk of damage every time a cell divides, because mechanisms that copy DNA and verify the copy during division aren't perfect. So old age alone, when DNA has been damaged in various types of cells that have divided too often, can also be a cause of cancer. Dangerous gene mutations can also be inherited, as already noted.
Mutations in genes that code for proteins that directly or indirectly promote cell division are, logically enough, one source of increased cancer risk. A mutation in some gene affecting a constituent of NF-κB would be an example here if, say, the mutation rendered NF-κB less subject to regulation by the proteins that normally regulate it. And as noted, mutations in genes for proteins that detect or repair DNA damage, or simply inhibit cell division when DNA damage exists, would also raise cancer risk. In both cases, the amount of risk also depends on environmental conditions, such as any that could cause inflammation. Mutations in many other genes can also affect cancer risk, when such genes are involved in angiogenesis or cell motility, for example.
Although a wide variety of DNA mutations can raise cancer risk, they may not be sufficient by themselves to actually initiate the development of cancer, because nature has provided cells with many defensive safety mechanisms. On the other hand, mutations aren't always necessary either. There are various external factors that might initiate cancer development even in the absence of DNA damage.
Frequently, it could be an infectious agent like a virus that stimulates excessive cell division. It's quite natural to expect some types of viruses to do this, because such viruses depend on a cell's normal DNA replication machinery to replicate virus DNA as well as cellular DNA. A virus that can easily co-opt the replication machinery has an evolutionary advantage. Especially if the virus can also override normal protective mechanisms, by inhibiting tumor-suppressing proteins like p53. All viruses bring along their own DNA or RNA, which may have evolved specifically because they disable anti-cancer mechanisms. Although virus genetic material is imported from outside, it acts like harmful gene mutations.
Viral replication strategies differ widely among different types of viruses, and certain of these strategies are especially conducive to cancer development. HPV, the human papilloma virus, which is responsible for cervical and anal cancers (among others) is an especially good example. Among the proteins that make up HPV are two, called E6 and E7, each of which promotes cell proliferation in its own way.
E6 is able to suppress the important anti-proliferation protein p53, which manages signals of DNA damage to take appropriate action, such as apoptosis or suspension of the cell cycle. E7 affects the protein pRb, which normally suppresses the cell cycle by binding to a transcription factor known as E2F. When E7 binds to pRb, E2F is released and can go on to advance the cell cycle, which then causes replication of HPV as well as unwanted cell division.
However, what HPV does isn't the only way that an infectious agent such as a virus can promote cancer. Infectious agents also activate the body's immune system to produce an inflammatory response. This inflammation itself can be a cause of cancer. Briefly stated, inflammation causes NF-κB to be activated in order to cause expression of genes that help invoke other immune system components to fight the infection. But NF-κB also has a side-effect of suppressing apoptosis, and as noted above, that is one of the "hallmarks" of cancer.
A connection between inflammation and cancer was suspected over 100 years ago by scientists like Rudolf Virchow. But only rather recently has solid evidence for the connection been found. A good example is Helicobacter pylori bacterial infections associated with stomach cancer (as well as stomach ulcers). A more recent example is an apparent link between inflammation, due to infection caused by the protozoan Trichomonas vaginalis, and prostate cancer. (See here.) Epidemiological evidence suggests that underlying infections and inflammation are associated with 15-20% of all cancer deaths.
There seem to be a number of factors that explain the connection. NF-κB seems to be one of the most important factors, though not the only one. It doesn't work only by suppression of apoptosis either. Promotion of angiogenesis, among other things, also seems to be involved. But most likely we still don't have a very complete understanding of the connection.
It's especially important to understand the connection, because infections aren't the only cause of inflammation. Other suspected causes of inflammation include stress and obesity. Understanding how obesity might promote cancer is obviously of no small importance. There is even evidence that depression may cause inflammation (see here), so that it could also lead to cancer.
In what follows, we're going to encounter various proteins and protein complexes that interact with each other in cell signaling pathways. Often this interaction takes the form that protein A inhibits the activity of protein B; while protein B inhibits the activity of protein C. The net effect is that protein A enhances the activity of protein C, and hence promotes any process that protein C assists in. Or if protein C inhibits some process, protein A will probably do likewise. This complexity can be very confusing, but it's also pretty common, so we just have to deal with it. In fact, the complexity of processes associated with cancer (and much other biology as well) is an important lesson in all of this.
Let's first consider the process of inflammation that occurs "upstream" from NF-κB and activates it. Inflammation refers to the whole process that occurs in a state of hightened immune system activity, due to physiological stress, oxidative stress, infection, or whatever. The outwards signs of inflammation include redness and swelling. There is a beneficial effect of inflammatory activity, of course, in (hopefully) destroying pathogens. But there are harmful side effects as well, including cardiovascular disease, diabetes, a variety of autoimmune diseases, and... cancer.
Many environmental stimuli lead to inflammation and so can cause NF-κB to be activated. Among these stimuli are stress, free radicals, ultraviolet irradiation, oxidized LDL (cholesterol), products of necrotic cell death, and bacterial or viral antigens.
Infectious agents such as bacteria and viruses contain proteins that act as antigens. These antigens are recognized by various cell-surface receptors, especially the kind known as Toll-like receptors (TLRs). Binding by appropriate proteins or antigens ("ligands") to such receptors is sufficient to activate NF-κB, in a manner we'll describe in a moment.
Antigens also bind to and stimulate cells of the immune system to produce various chemical signals such as cytokines, chemokines, and other proteins in order to regulate the immune system response to infection. Among the types of immune system cells that do this are mast cells, dendritic cells, neutrophils, eosinophils, macrophages, and various other lymphocytes. Inflammatory cytokines can also cause activation of NF-κB.
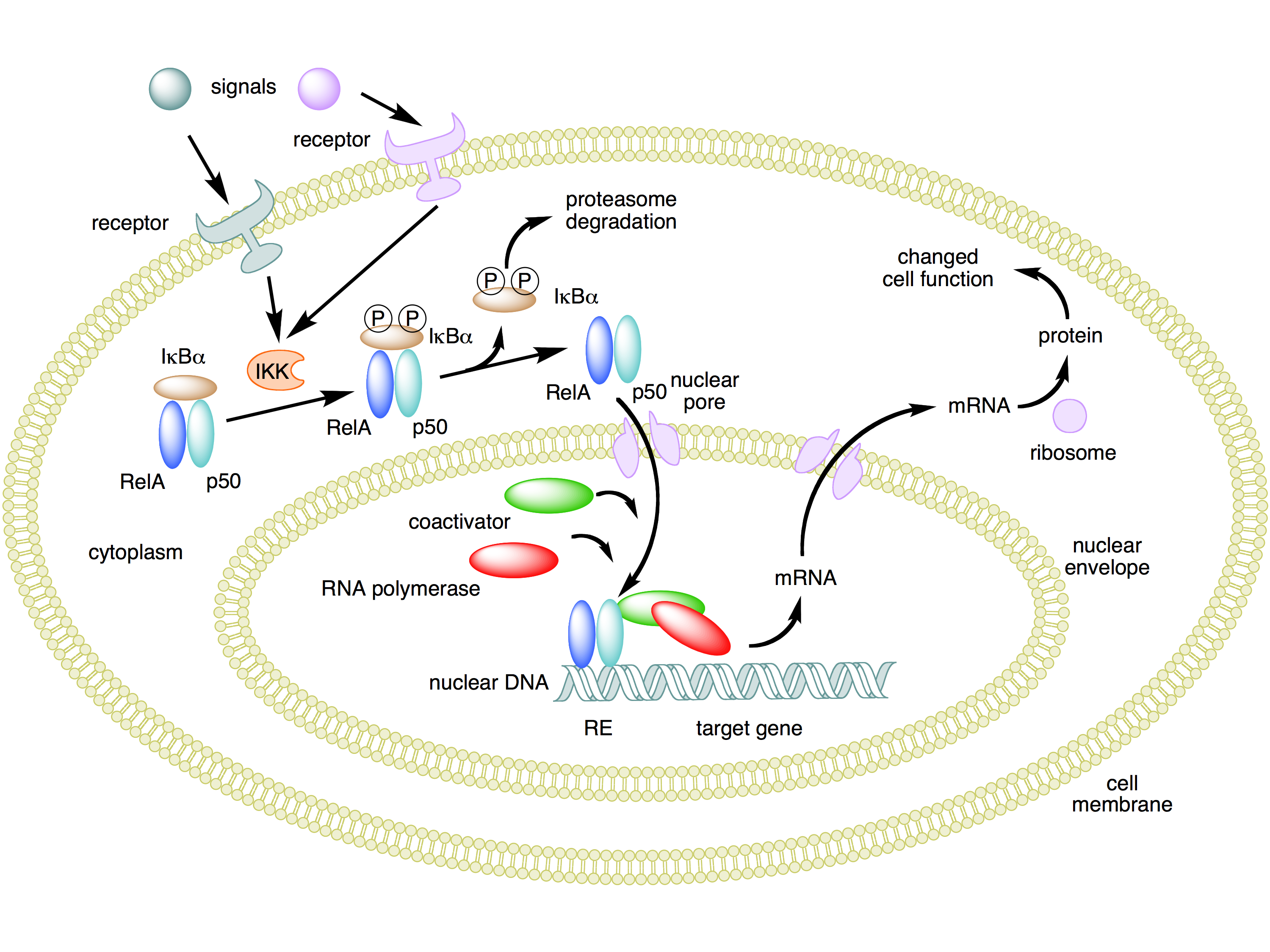
The crucial effect of inflammation for our purposes now is the fact that it activates NF-κB. It's worth noting how this process works. NF-κB is a protein complex consisting of five protein subunits, not a single protein. These subunits are RelA (also known as p65), c-Rel, RelB, p50, and p52. Complexes of these subunits normally circulate outside the cell nucleus, but they are bound to other proteins called IκBs ("inhibitors of NF-κBs") that prevent the complexes from entering the cell nucleus where they could act as transcription factors.
When an inflammatory signal binds to an appropriate cell surface receptor, signals are sent that activate kinases of a family called IKK (IκB kinase). An IKK protein phosphorylates IκBs, which in turn causes them to become unbound from NF-κB and then be destroyed by cellular proteasomes. This frees up NF-κB complexes so that they can enter the cell nucleus and affect the transcription of many genes.
The benefit of an inhibited form of NF-κB existing in the cytoplasm outside the nucleus is that it can be quickly enabled to enter the nucleus and start gene transcription when the need arises. This allows NF-κB to function as a "rapid-acting" transcription factor, without any delays caused by having to wait for the constituent proteins to be synthesized. Here's a diagram that summarizes this process. (Similar considerations apply to p53. The protein is produced and is found in the cytoplasm before it's needed, but kept from activity while bound to another protein, Mdm2.)
The next issue is what happens downstream from the inflammation-initiated activity of NF-κB in the nucleus. Since NF-κB can assist in the transcription of many different genes, the effect of its activation strongly depends on what type of cell it occurs in. Undoubtedly, there's a whole lot we don't yet know about all the affected genes and resulting downstream effects. As far as the immune response – in which NF-κB plays such an important role – is concerned, one effect involves production of cytokines for signaling to other immune system cells.
A second effect is stimulation of cell proliferation. That's generally a good thing if the cell is, for example, a B cell that mediates the part of the immune respose by manufacturing antibodies. But it can also be a bad thing when excessive proliferation of B cells leads to autoimmune diseases, leukemias, or lymphomas. NF-κB stimulates proliferation by enhancing expression of cell cycle proteins like cyclin D1.
However, there's also a third type of effect of NF-κB activity – inhibition of apoptosis. This is especially significant for cancer, because apoptosis is crucial for many natural anti-cancer cellular defenses. In particular, apoptosis is the normal response to severe, uncorrectable DNA damage. It's also the typical way that chemotherapy is able to kill tumor cells. Interference with normal apoptosis makes cancer both more likely to occur, and more difficult to treat.
Exactly how does NF-κB activity inhibit apoptosis? This has been studied, and the answer seems to be that NF-κB inhibits certain enzymes called caspases that are central to apoptosis.
There are even further suspected side effects of NF-κB activity which can play a big role in cancer. One of these is promotion of angiogenesis – production of blood vessels that can supply nutrients to solid tumors. Another is elevated expression of enzymes that promote metastasis.
Observe that the discussion here has been largely theoretical. We haven't described actual experimental research that supports the generalizations. There is some recent research already mentioned that fills this gap. It's especially interesting in the way it exposes a direct connection between NF-κB and p53. But we must leave description of that research for another article, coming very soon.
Further reading:
Nuclear factor-κB in cancer development and progression – May 2006 Nature review article
Tags: cancer, inflammation
At the very center of this story is a transcription factor called NF-κB. It's perhaps best known for its role in inflammation. But we discussed it in connection with cancer over two years ago, here. Since NF-κB is a transcription factor it affects the expression of many genes. Consequently, it's involved in many other biological phenomena, a few of which have been discussed here, here, and here.
Let's review some of the characteristics shared by most forms of cancer. One frequently cited summary, due to Douglas Hanahan and Robert Weinberg, names six "hallmarks of cancer" – self-sufficiency in growth signals, insensitivity to growth-inhibitory signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis. It turns out that inflammation and NF-κB can affect most of these hallmarks, but we'll eventually focus on the role NF-κB plays in inhibition of apoptosis (programmed cell death).
Before we get into details, let's go over the way that cancer typically develops. Each of the hallmarks represents a failure in one or more parts of the machinery of cells to properly regulate the cell's life cycle. The bad thing about cancer, of course, is that a certain group of cells – which might all be descendants of a single aberrant cell – begins to grow and proliferate in an unregulated way. Eventually large numbers of unregulated cells will cease to function as required in the organ where they reside – the brain, the liver, breast tissue, or whatever. Instead they will acquire the ability to migrate and take up residence in places where they continue to proliferate and then disrupt the proper function of whatever organ they wind up in. This is metastasis, and it is usually fatal to the animal in which it occurs.
The cell's machinery is ultimately controlled by the way specific genes of the cell's DNA are expressed to produce proteins. So the reason a cell initially becomes improperly regulated is usually the occurrence of fresh damage to the cell's DNA. It's true that problematic DNA mutations can be inherited from parents, such as the well-known breast cancer genes BRCA1 and BRCA2. Signifcantly, the normal function of both these genes, when not mutated, is to produce proteins whose job is to detect and/or repair DNA damage or to arrest cell proliferation if DNA damage is detected but can't be repaired. So mutated versions of these genes do not exactly "cause" cancer themselves, since some mutation to other genes that actively induce excessive cell proliferation is also required. The potential for cancer may simply remains latent for awhile in cells with mutated genes, regardless of whether the mutation was inherited or occurred much later in life.
But eventually uncorrected DNA damage will occur and affect genes that do induce excessive proliferation. Metastatic cancer eventually results, regardless of whether the first harmful mutations occur in genes that induce proliferation or genes that help regulate proliferation. Cells that contain such mutations divide more frequently and outcompete for resources cells that are functioning properly. So a Darwinian evolutionary scenario arises in which cells that are working "correctly" lose out to cells that become better "adapted" to the job of simply replicating themselves.
The point of this general discussion about cancer is to help clarify what it means to say that something "causes" cancer. In reality, there's never a single event that is the "cause". A number of things have to go wrong before some population of cells with similar defects is numerous enough to outcompete properly functioning cells and to go on to acquire further defects when the mechanisms that normally protect against damage themselves begin to fail.
There appear to be several ways in which NF-κB can contribute to the development of cancer, but one of the least surprising ways is the fact that NF-κB is able to inhibit apoptosis. This ability is not accidental. Most likely it is properly there to enable proliferation of immune system cells in the presence of an infection, as indicated by inflammatory signals.
To return to our main topic, inflammation is a thoroughly normal part of the immune system's operation. NF-κB plays an important part in the inflammatory process. But in simply doing their jobs, these things can, under the right conditions when other cellular mechanisms befome defective, also contribute to the development and progress of cancer.
Normally, the ability of NF-κB to inhibit apoptosis doesn't present a risk of cancer because NF-κB is regulated by other proteins, as we'll discuss later. However, if there are gene mutations that affect NF-κB regulatory proteins, the "safety catch" mechanism may be compromised, and apoptosis may be inhibited when it shouldn't be. Such a problem is more likely to occur when inflammation is present, since then the "safety catch" is already partly disabled.
Another mechanism that keeps NF-κB inhibition of apoptosis in check is the existence of pro-apoptotic proteins such as p53. (We've discussed p53 a number of times before, most recently here, where new research about the anti-cancer properties of p53 is described.) Such proteins are normally subject to regulation themselves, and they become available and active only when needed, such as when DNA damage is detected. However, when one of these proteins has itself been compromised by a gene mutation, inflammation and resultant NF-κB activity can inappropriately inhibit apoptosis, because pro-apoptotic factors are weakened or sidelined.
As it turns out, according to recent research, among the ways that p53 promotes apoptosis is by direct interference with NF-κB's ability to inhibit apoptosis. Further, certain mutations of the p53 gene can remove p53's pro-apoptotic ability, allowing inflammation and NF-κB to contribute to development of cancer. (See here. We'll discuss that in a separate article.)
The high-level view of all this is that cells exist continually in a state of balance between opposing possibilities. Pro-apoptotic and anti-apoptotic mechanisms are not only regulated independently, but they also keep each other in check. But when mutations in any number of possible genes upset the balance, otherwise normal and useful mechanisms can lead to cancer.
Now let's go a little deeper into the subject of various factors that actively contribute to cancer, starting with DNA damage – mutations. The class of things that cause mutations comprises factors such as carcinogenic chemicals, reactive oxygen species ("free radicals"), ultraviolet light, ionizing radiation (e. g. x-rays, radon gas), and some types of viruses. DNA is also at risk of damage every time a cell divides, because mechanisms that copy DNA and verify the copy during division aren't perfect. So old age alone, when DNA has been damaged in various types of cells that have divided too often, can also be a cause of cancer. Dangerous gene mutations can also be inherited, as already noted.
Mutations in genes that code for proteins that directly or indirectly promote cell division are, logically enough, one source of increased cancer risk. A mutation in some gene affecting a constituent of NF-κB would be an example here if, say, the mutation rendered NF-κB less subject to regulation by the proteins that normally regulate it. And as noted, mutations in genes for proteins that detect or repair DNA damage, or simply inhibit cell division when DNA damage exists, would also raise cancer risk. In both cases, the amount of risk also depends on environmental conditions, such as any that could cause inflammation. Mutations in many other genes can also affect cancer risk, when such genes are involved in angiogenesis or cell motility, for example.
Although a wide variety of DNA mutations can raise cancer risk, they may not be sufficient by themselves to actually initiate the development of cancer, because nature has provided cells with many defensive safety mechanisms. On the other hand, mutations aren't always necessary either. There are various external factors that might initiate cancer development even in the absence of DNA damage.
Frequently, it could be an infectious agent like a virus that stimulates excessive cell division. It's quite natural to expect some types of viruses to do this, because such viruses depend on a cell's normal DNA replication machinery to replicate virus DNA as well as cellular DNA. A virus that can easily co-opt the replication machinery has an evolutionary advantage. Especially if the virus can also override normal protective mechanisms, by inhibiting tumor-suppressing proteins like p53. All viruses bring along their own DNA or RNA, which may have evolved specifically because they disable anti-cancer mechanisms. Although virus genetic material is imported from outside, it acts like harmful gene mutations.
Viral replication strategies differ widely among different types of viruses, and certain of these strategies are especially conducive to cancer development. HPV, the human papilloma virus, which is responsible for cervical and anal cancers (among others) is an especially good example. Among the proteins that make up HPV are two, called E6 and E7, each of which promotes cell proliferation in its own way.
E6 is able to suppress the important anti-proliferation protein p53, which manages signals of DNA damage to take appropriate action, such as apoptosis or suspension of the cell cycle. E7 affects the protein pRb, which normally suppresses the cell cycle by binding to a transcription factor known as E2F. When E7 binds to pRb, E2F is released and can go on to advance the cell cycle, which then causes replication of HPV as well as unwanted cell division.
However, what HPV does isn't the only way that an infectious agent such as a virus can promote cancer. Infectious agents also activate the body's immune system to produce an inflammatory response. This inflammation itself can be a cause of cancer. Briefly stated, inflammation causes NF-κB to be activated in order to cause expression of genes that help invoke other immune system components to fight the infection. But NF-κB also has a side-effect of suppressing apoptosis, and as noted above, that is one of the "hallmarks" of cancer.
A connection between inflammation and cancer was suspected over 100 years ago by scientists like Rudolf Virchow. But only rather recently has solid evidence for the connection been found. A good example is Helicobacter pylori bacterial infections associated with stomach cancer (as well as stomach ulcers). A more recent example is an apparent link between inflammation, due to infection caused by the protozoan Trichomonas vaginalis, and prostate cancer. (See here.) Epidemiological evidence suggests that underlying infections and inflammation are associated with 15-20% of all cancer deaths.
There seem to be a number of factors that explain the connection. NF-κB seems to be one of the most important factors, though not the only one. It doesn't work only by suppression of apoptosis either. Promotion of angiogenesis, among other things, also seems to be involved. But most likely we still don't have a very complete understanding of the connection.
It's especially important to understand the connection, because infections aren't the only cause of inflammation. Other suspected causes of inflammation include stress and obesity. Understanding how obesity might promote cancer is obviously of no small importance. There is even evidence that depression may cause inflammation (see here), so that it could also lead to cancer.
In what follows, we're going to encounter various proteins and protein complexes that interact with each other in cell signaling pathways. Often this interaction takes the form that protein A inhibits the activity of protein B; while protein B inhibits the activity of protein C. The net effect is that protein A enhances the activity of protein C, and hence promotes any process that protein C assists in. Or if protein C inhibits some process, protein A will probably do likewise. This complexity can be very confusing, but it's also pretty common, so we just have to deal with it. In fact, the complexity of processes associated with cancer (and much other biology as well) is an important lesson in all of this.
Let's first consider the process of inflammation that occurs "upstream" from NF-κB and activates it. Inflammation refers to the whole process that occurs in a state of hightened immune system activity, due to physiological stress, oxidative stress, infection, or whatever. The outwards signs of inflammation include redness and swelling. There is a beneficial effect of inflammatory activity, of course, in (hopefully) destroying pathogens. But there are harmful side effects as well, including cardiovascular disease, diabetes, a variety of autoimmune diseases, and... cancer.
Many environmental stimuli lead to inflammation and so can cause NF-κB to be activated. Among these stimuli are stress, free radicals, ultraviolet irradiation, oxidized LDL (cholesterol), products of necrotic cell death, and bacterial or viral antigens.
Infectious agents such as bacteria and viruses contain proteins that act as antigens. These antigens are recognized by various cell-surface receptors, especially the kind known as Toll-like receptors (TLRs). Binding by appropriate proteins or antigens ("ligands") to such receptors is sufficient to activate NF-κB, in a manner we'll describe in a moment.
Antigens also bind to and stimulate cells of the immune system to produce various chemical signals such as cytokines, chemokines, and other proteins in order to regulate the immune system response to infection. Among the types of immune system cells that do this are mast cells, dendritic cells, neutrophils, eosinophils, macrophages, and various other lymphocytes. Inflammatory cytokines can also cause activation of NF-κB.
The crucial effect of inflammation for our purposes now is the fact that it activates NF-κB. It's worth noting how this process works. NF-κB is a protein complex consisting of five protein subunits, not a single protein. These subunits are RelA (also known as p65), c-Rel, RelB, p50, and p52. Complexes of these subunits normally circulate outside the cell nucleus, but they are bound to other proteins called IκBs ("inhibitors of NF-κBs") that prevent the complexes from entering the cell nucleus where they could act as transcription factors.
When an inflammatory signal binds to an appropriate cell surface receptor, signals are sent that activate kinases of a family called IKK (IκB kinase). An IKK protein phosphorylates IκBs, which in turn causes them to become unbound from NF-κB and then be destroyed by cellular proteasomes. This frees up NF-κB complexes so that they can enter the cell nucleus and affect the transcription of many genes.
The benefit of an inhibited form of NF-κB existing in the cytoplasm outside the nucleus is that it can be quickly enabled to enter the nucleus and start gene transcription when the need arises. This allows NF-κB to function as a "rapid-acting" transcription factor, without any delays caused by having to wait for the constituent proteins to be synthesized. Here's a diagram that summarizes this process. (Similar considerations apply to p53. The protein is produced and is found in the cytoplasm before it's needed, but kept from activity while bound to another protein, Mdm2.)
The next issue is what happens downstream from the inflammation-initiated activity of NF-κB in the nucleus. Since NF-κB can assist in the transcription of many different genes, the effect of its activation strongly depends on what type of cell it occurs in. Undoubtedly, there's a whole lot we don't yet know about all the affected genes and resulting downstream effects. As far as the immune response – in which NF-κB plays such an important role – is concerned, one effect involves production of cytokines for signaling to other immune system cells.
A second effect is stimulation of cell proliferation. That's generally a good thing if the cell is, for example, a B cell that mediates the part of the immune respose by manufacturing antibodies. But it can also be a bad thing when excessive proliferation of B cells leads to autoimmune diseases, leukemias, or lymphomas. NF-κB stimulates proliferation by enhancing expression of cell cycle proteins like cyclin D1.
However, there's also a third type of effect of NF-κB activity – inhibition of apoptosis. This is especially significant for cancer, because apoptosis is crucial for many natural anti-cancer cellular defenses. In particular, apoptosis is the normal response to severe, uncorrectable DNA damage. It's also the typical way that chemotherapy is able to kill tumor cells. Interference with normal apoptosis makes cancer both more likely to occur, and more difficult to treat.
Exactly how does NF-κB activity inhibit apoptosis? This has been studied, and the answer seems to be that NF-κB inhibits certain enzymes called caspases that are central to apoptosis.
There are even further suspected side effects of NF-κB activity which can play a big role in cancer. One of these is promotion of angiogenesis – production of blood vessels that can supply nutrients to solid tumors. Another is elevated expression of enzymes that promote metastasis.
Observe that the discussion here has been largely theoretical. We haven't described actual experimental research that supports the generalizations. There is some recent research already mentioned that fills this gap. It's especially interesting in the way it exposes a direct connection between NF-κB and p53. But we must leave description of that research for another article, coming very soon.
Further reading:
Nuclear factor-κB in cancer development and progression – May 2006 Nature review article
Tags: cancer, inflammation
Labels: cancer, cancer biology, inflammation, NF-kB

 Subscribe to posts
Subscribe to posts

 Google
Google Delicious
Delicious Diigo
Diigo Twine
Twine StumbleUpon
StumbleUpon







{kind=link}
{kind=link}